Introduction
ChIP-Seq is widely used to characterize genome-wide binding patterns of transcription factors and other chromatin-associated proteins. Although comparison of ChIP-Seq data sets is critical for understanding cell type-dependent and cell state-specific binding, and thus the study of cell-specific gene regulation, few quantitative approaches have been developed. Here, we present a simple and effective method, MAnorm, for quantitative comparison of ChIP-Seq data sets describing transcription factor binding sites and epigenetic modifications. The quantitative binding differences inferred by MAnorm showed strong correlation with both the changes in expression of target genes and the binding of cell type-specific regulators.
Reference
Downloads
| R version: | download | tutorial | contact |
| MATLAB version: | download | tutorial | contact |
Model Description
1. AssumptionsFirst, we assume the true intensities of most common peaks are the same between two ChIP-Seq samples. This assumption is valid when the binding regions represented by the common peaks show a much higher level of co-localization between samples than that expected at random, and thus binding at the common peaks should be determined by similar mechanisms and exhibit similar global binding intensity between samples.
Second, the observed differences in sequence read density in common peaks are presumed to reflect the scaling relationship of ChIP-Seq signals between two samples, which can thus be applied to all peaks.
2. Workflow
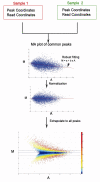
Further Applications
1. Quantitative comparison of binding difference with target gene expression change. 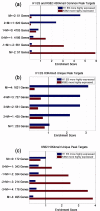
2. Searching for potential cell line-specific co-factors that contribute to differential binding.

Acknowledgements
We thank Aarathi Sugathan (Boston University) for sharing ideas and scripts during MAnorm BASH/R package development; we also thank Andy Rampersaud (Boston University), Drs. Jian Xu and Han Xu (Dana-Farber Cancer Institute) for many useful discussions and suggestions during the course of this study.