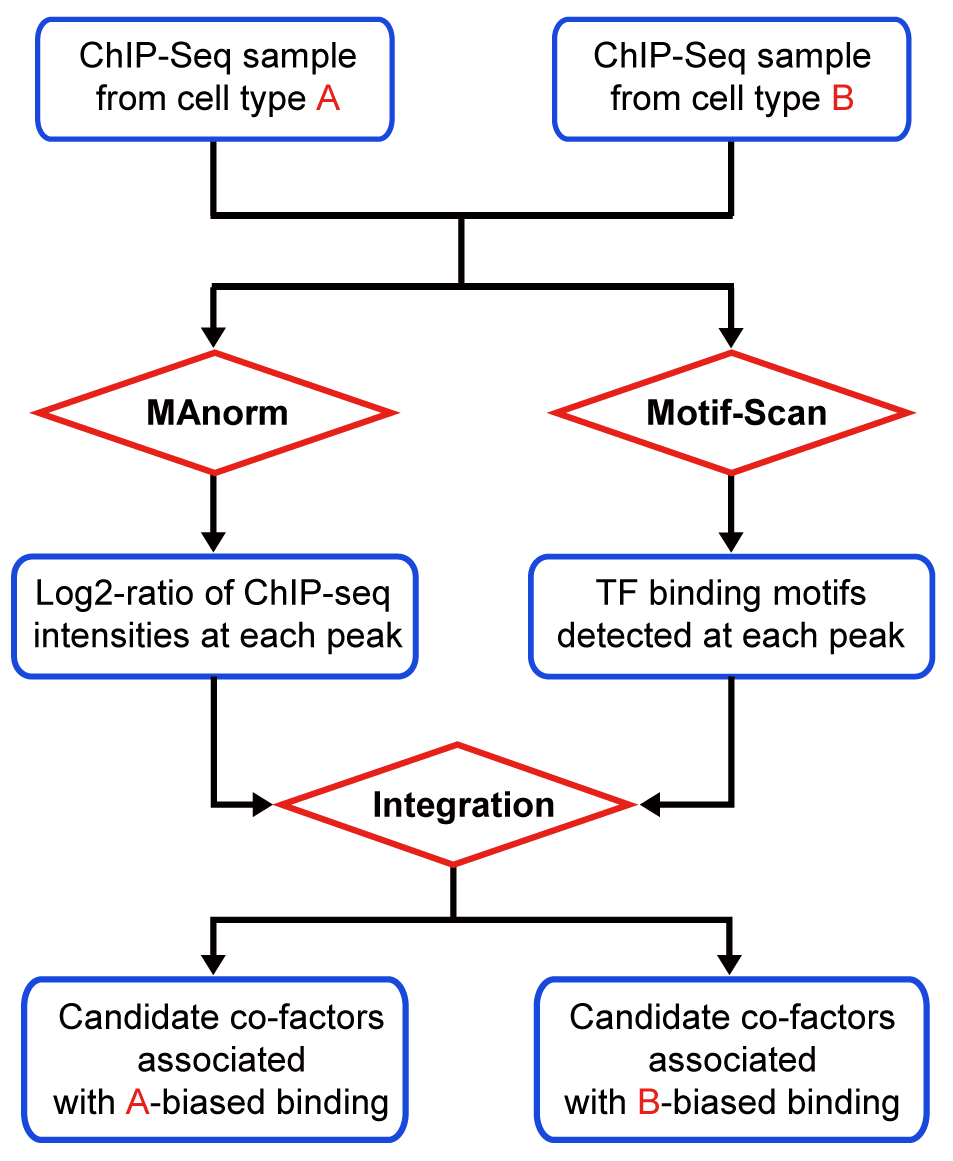
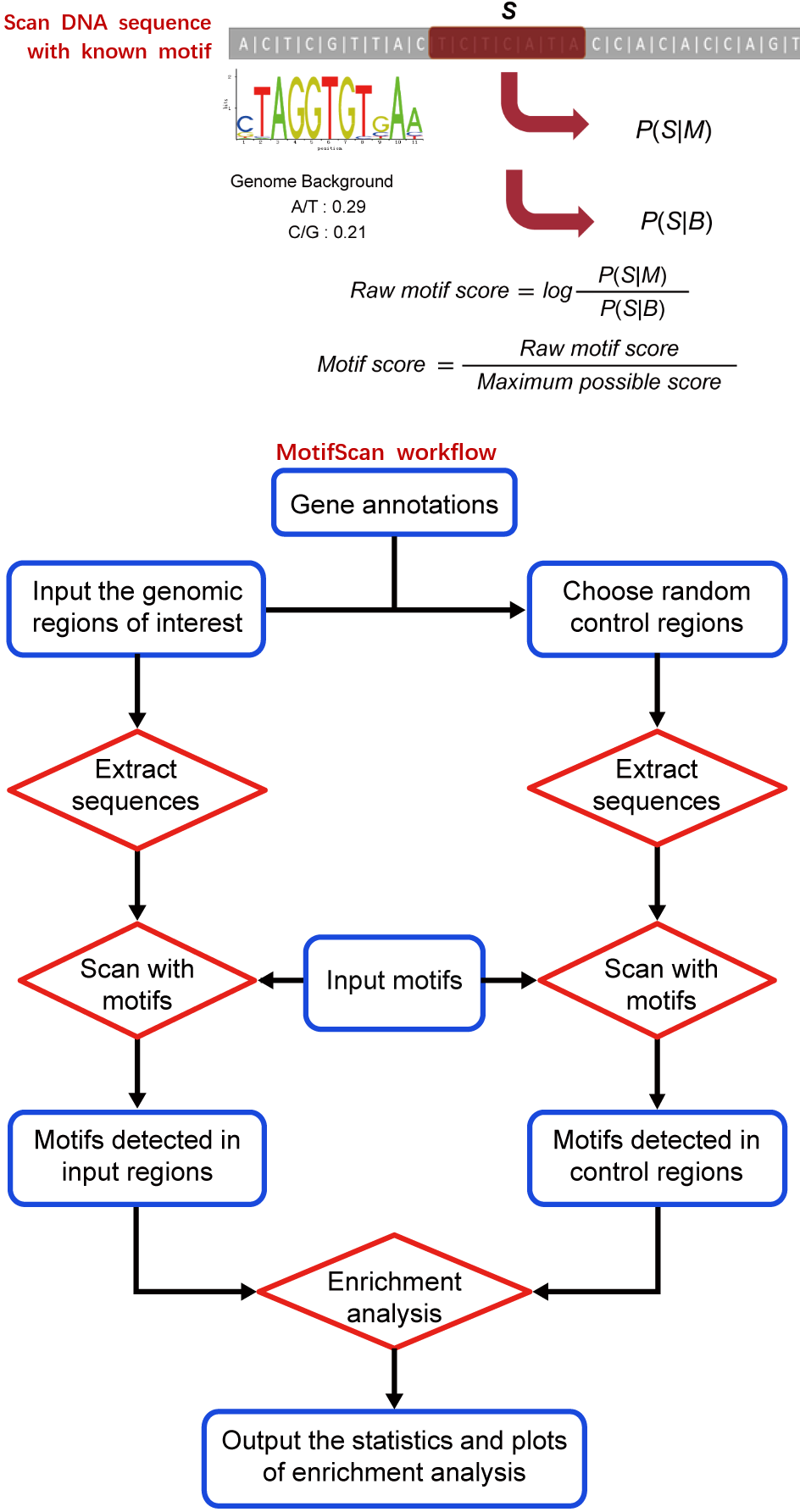
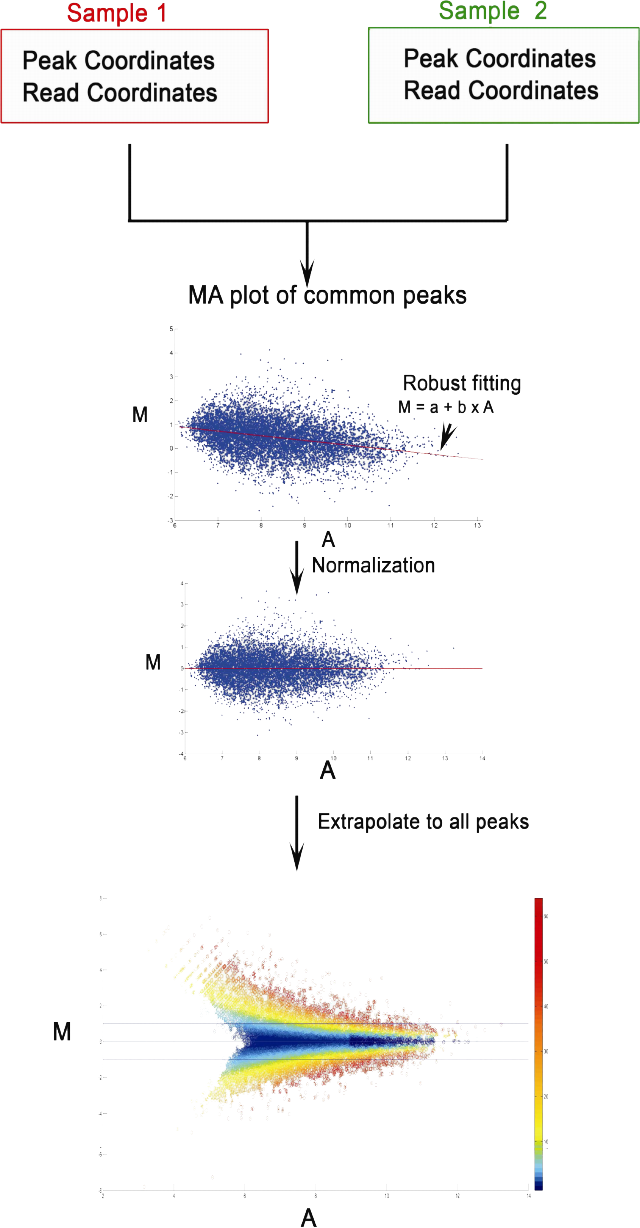
MAnorm2 for quantitatively comparing groups of ChIP/ATAC/DNase-seq samples

Email: shaozhen@picb.ac.cn
Office: Shengli Building 330
Tel. number: 021-54920367
Education and Work Experience:
From 2021.1: Group Leader, CAS Key Lab of Computational Biology, Shanghai Institute of Nutrition and Health, CAS
2013.10 - 2020.12: Group Leader, CAS-MPG Partner Institute for Computational Biology, Shanghai Institute of Nutrition and Health (formed in 2017.1), Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences
2010.6 - 2013.9: Postdoctoral Fellow, Department of Pediatric Oncology, Dana Farber Cancer Institute; Division of Hematology/Oncology, Children’s Hospital Boston; Harvard Medical School;
2009.4 - 2010.5: Postdoctoral Fellow, Department of Biostatistics and Computational Biology, Dana Farber Cancer Institute and Department of Biostatistics, Harvard School of Public Health;
2003.9 - 2008.12: Ph.D. of Theoretical Biophysics, Institute of Theoretical Physics, Chinese Academy of Sciences, China;
1999.9 - 2003.7: Bachelor of Theoretical Physics, University of Science and Technology of China.